In pharma, speed and compliance are everything. Yet behind every approved drug lies a lesser-known tool: the Drug Master File (DMF).
DMFs let companies share critical data with the FDA, without revealing trade secrets. It’s not legally required, but skipping it can stall or derail approvals.
Used in NDAs, ANDAs, and INDs, DMFs help safeguard intellectual property while fast-tracking regulatory review.
This guide breaks down what DMFs are, why they matter, and how they quietly drive drug approvals forward.
What is a Drug Master File (DMF)?
A Drug Master File (DMF) acts like a behind-the-scenes dossier. It quietly powers many drug approvals without stepping into the spotlight.
Pharmaceutical manufacturers submit DMFs to the US FDA. These documents contain sensitive information about facilities, ingredients, processes, and quality controls used in drug manufacturing.
Rather than expose trade secrets, companies can keep critical data confidential. At the same time, the FDA gets what it needs to evaluate a drug's safety and quality. This information is not made public. It isn’t required by law either. But in most cases, skipping a DMF would slow your approval process—or stop it completely.
DMFs are reviewed during the approval of NDAs, ANDAs, and INDs. They’re never approved or rejected themselves, but their contents are closely examined. By using DMFs, companies protect intellectual property while staying compliant. It’s a quiet move—but a powerful one.
Types of Drug Master Files
Not all DMFs are created equal. Each type serves a specific function, depending on what you're trying to submit or protect.
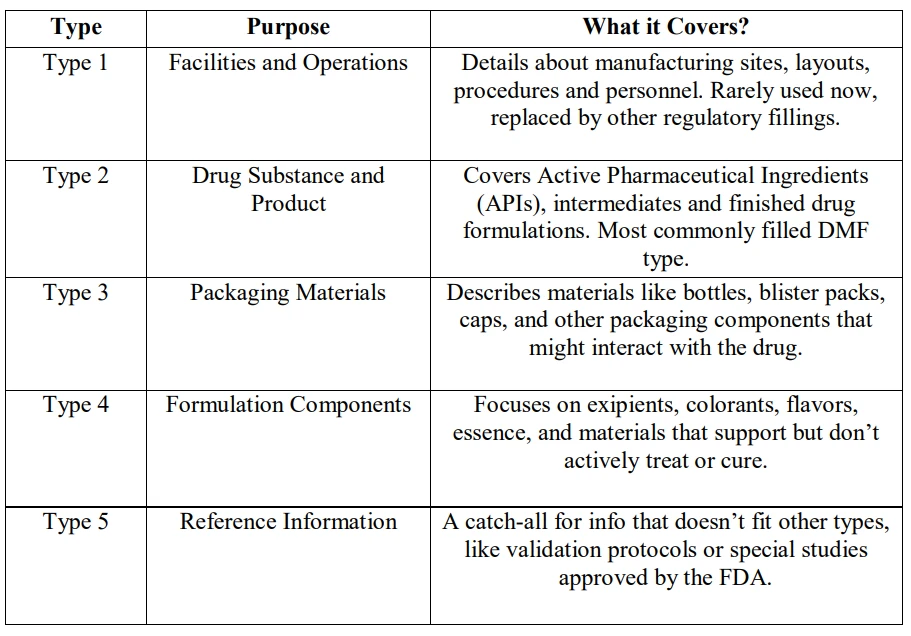
Each DMF type exists for a reason. They help companies submit precise data while keeping trade secrets safe, and help regulators make smarter, faster decisions.
The Role of DMFs in Regulatory Compliance and Certifications
One of the major role of DMF in regulatoryc compliance and certifications is that it protects proprietary information and smooths your path through regulatory checkpoints.
While not legally required, DMFs are widely adopted in the pharmaceutical industry. Why? Because they offer significant strategic advantages.
Here’s how DMFs support compliance and certification:
Confidential but compliant: DMFs allow manufacturers to submit sensitive technical data to the FDA without sharing it with clients or competitors.
Trusted by regulators: The FDA uses DMFs to evaluate the quality, safety, and integrity of drug substances and components.
Streamlines approvals: By referencing a DMF, companies can reduce duplication in NDA or ANDA filings.
Supports cGMP adherence: DMFs reinforce compliance with current Good Manufacturing Practices by detailing materials, methods, and quality controls.
Protects IP while aiding collaboration: Suppliers can provide essential data to clients via FDA reference—without exposing proprietary content.
For pharma professionals, submitting a DMF communicates three key messages:
“We understand the regulations.”
"We operate with transparency.”
“We’re committed to high standards.”
In short, a well-maintained DMF isn’t just a document, it’s a mark of credibility.
Preparing a DMF: Key Considerations
Preparing a Drug Master File isn’t just about paperwork. It’s a disciplined process that reflects your operations, controls, and regulatory readiness.
Here’s what you need to know:
Documentation Requirements
You must include detailed descriptions of facilities, materials, processes, and testing methods.
Clarity matters. The FDA must easily verify your compliance with cGMP standards.
Each section must follow the Common Technical Document (CTD) format as recommended.
Incomplete or vague sections often trigger review delays or FDA queries.
Tips for a Successful Submission
Be precise, not verbose. Avoid jargon. Use specific terms to describe processes and controls.
Review regularly. Keep documentation updated to reflect real-time changes in materials or methods.
Cross-check for accuracy. Small errors in data or terminology can create big setbacks.
Have experts review it. A second pair of trained eyes can save months of back-and-forth with regulators.
Version control matters. Always submit the latest, approved versions of your SOPs and batch records.
Success isn’t luck, it’s preparation meeting the right process. Your DMF should speak clearly, comply fully, and anticipate questions before they’re asked.
The Review Process of DMFs
Submitting a DMF is just the beginning. What happens next determines how smoothly your drug application progresses.
Steps Involved in DMF Review
Initial Acknowledgment: Once submitted, the FDA issues an acknowledgment letter confirming receipt.
Administrative Review: The FDA checks for format, completeness, and compliance with submission standards.
Technical Review: Subject-matter experts review the data—checking safety, quality, and compliance with cGMP.
Deficiency Letter (if needed): If gaps or errors are found, a letter is sent requesting additional information.
Final Decision: Upon successful review, the DMF is accepted and may be referenced in future applications.
Each review step demands clarity, precision, and prompt follow-up from the submitting company.
Conclusion
Drug Master Files aren’t just paperwork. They’re powerful regulatory tools that protect intellectual property while accelerating drug approvals and ensuring product quality.
Understanding DMFs means knowing what regulators look for, how information flows confidentially, and where your compliance efforts should focus. Whether you’re a manufacturer, API buyers & Supplier , or generic drug developer — mastering the DMF process gives you a strategic edge in pharma.
Keep your documentation tight. Respond quickly to FDA queries. And always update your DMF when changes occur, this isn’t a one-and-done document. As the pharmaceutical landscape evolves, DMFs will play an even greater role in global regulatory harmonization and cross-border approvals. Get familiar, stay proactive, and treat DMFs not as a burden, but as a gateway to regulatory trust and long-term market access.