Complete Process DMPK Service Helps PROTAC Drug Development
To date, there are few literature reports that summarize PROTAC metabolism studies. Therefore, in this article, we share some examples of DMPK studies of PROTAC with the experience of the Medicilon DMPK department and hope to be helpful to all researchers who are interested in PROTAC molecular DMPK.
Prostate cancer is a common malignancy in men, and the development of effective therapies for prostate cancer has been a primary focus of scientific research. The prostate is an androgen-dependent organ, and prostate cancer is an androgen-dependent disease. Androgen action is mediated by the androgen receptor (AR), a hormone-activated transcription factor that belongs to the nuclear receptor superfamily of steroid receptors. AR and its downstream signaling play a vital role in the development and progression of localized and metastatic prostate cancer.
 structure.webp")
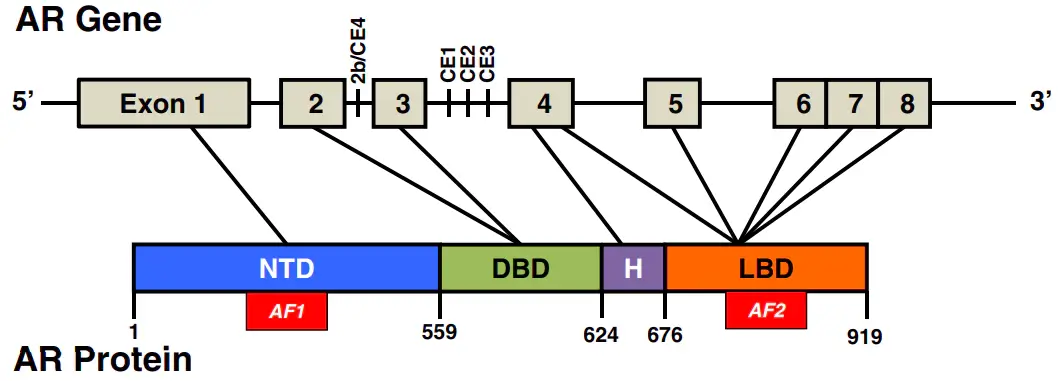
Androgen receptor (AR) structure [1]
The primary treatment for metastatic prostate cancer is androgen deprivation therapy (ADT). Many drugs targeting the AR have been developed to treat advanced prostate cancer, including Abiraterone, which blocks androgen synthesis, and the AR antagonists Enzalutamide, Apalutamide, and Darolutamide, which bind to the AR. However, resistance to these drugs usually develops within two years of treatment. In most tumors resistant to AR antagonists, AR signaling continues to function and drive tumor growth and progression. Resistant prostate cancer (CRPC) is dependent on AR. AR gene amplification, point mutations, and selective splicing have been identified as some of the primary mechanisms underlying resistance to these drugs targeting AR.
In addition, changes in steroid metabolism, cellular signaling, and co-regulatory proteins are also essential factors in AR reactivation in CRPC. Most AR-targeted therapies target hormone-binding domains. Constitutively active AR splice variants lacking hormone-binding structural domains are frequently expressed in CRPC, a finding that underscores the need to develop therapies targeting other parts of AR. Therefore, there is an urgent need to develop new therapeutic strategies for treating prostate cancer with AR, especially for metastatic debulking-resistant prostate cancer (mCRPC). mCRPC remains incurable and fatal until now.
Targeted degradation of the androgen receptor (AR)
Inducible degradation of AR proteins may be more effective than traditional AR antagonists in targeting AR signaling. Selective androgen receptor degraders (SARDs) bind to the ligand-binding structural domain in AR and disrupt the interaction of AR with co-regulators, leading to proteasome-dependent AR degradation. Another new strategy for induced AR degradation is based on the Protein Hydrolysis Targeted Chimera (PROTAC) technology platform. PROTAC-based AR degraders are small bifunctional molecules consisting of an AR ligand bound to an AR protein and a ligand bound to an E3 ligase complex and linked together by a Linker. PROTAC small molecule degraders have emerged as promising new therapeutic agents, but designing PROTAC degraders with excellent oral pharmacokinetics is a significant challenge. Researchers have developed and synthesized efficient PROTAC AR degraders with high oral bioavailability in several studies based on novel strategies.
PROTAC AR degradation agent ARD-2128
In the following paper, the researchers used Cereblon/cullin 4A E3 ligase raised by Thalidomide and cured by Linker to discover ARD-2128, a highly effective AR degradation agent with good oral pharmacokinetic properties in mice.

In this study, researchers evaluated the pharmacokinetic (PK) data of five potent AR reducers in mice administered intravenously and orally by Medicilon, with ARD-2128 being the optimal compound. PK data showed that ARD-2128 had an excellent overall PK profile: low clearance (1.2 mL/min/kg) and moderate to the high steady-state volume of distribution (Vss of 2.7 L/kg). The T1/2 of ARD-2128 was 27.6 hours after intravenous administration at 2 mg/kg and 18.8 hours after oral administration at 5 mg/kg. ARD-2128 (5 mg/kg) achieved 67% oral bioavailability in mice. It effectively reduced AR protein and inhibited AR-regulated genes in tumor tissues by oral administration, resulting in effective inhibition of tumor growth in mice with no signs of toxicity.

Summary of PK data of five compounds in male ICR mice [2]
In addition, the researchers tested the plasma stability of ARD-2128 in five genera: mouse, rat, canine, monkey, and human by Medicilon. The data showed that ARD-2128 had stable plasma stability in all five genera.

Plasma stability of ARD-2128 in five genera[2]
PROTAC AR degradant ARD-2585
In the following section, researchers report some efficient and orally bioavailable PROTAC AR degraders, among which ARD-2585 is one of the most promising. ARD-2585 is an orally effective PROTAC AR degradation agent. ARD-2585 achieves DC50 values of ≤0.1 nM in VCaP cell lines with AR gene amplification and LNCaP cell lines carrying AR mutations. ARD-2585 effectively inhibited the growth of VCaP and LNCaP cells with IC50 values of 1.5 and 16.2 nM, respectively, and showed excellent pharmacokinetics and oral bioavailability (51%) in mice. ARD-2585 was more effective than Enzalutamide in inhibiting VCaP tumor growth and did not cause any signs of toxicity in mice. Therefore, ARD-2585 is a promising AR reducer for the treatment of advanced prostate cancer.

Researchers investigated the mechanism of AR degradation induced by ARD-2585 in VCaP and LNCaP cell lines and found that AR protein was effectively reduced by AR-2585 (100 nM) in VCaP and LNCaP cells after 3 hours of treatment. Pretreatment with AR inhibitor, Cereblon ligand Thalidomide, proteasome inhibitor MG-132, and E1 Neddylation inhibitor MLN4924 effectively blocked AR degradation. These data provide clear mechanistic evidence that ARD-2585 induces AR degradation via Cereblon, proteasome, and neddylation-dependent mechanisms and is, therefore, a bona fide PROTAC AR degradation agent.

Mechanism of action of ARD-2585 [3]
Researchers next tested the antitumor activity of ARD-2585 in a VCaP xenograft tumor model with Enzalutamide as the control. ARD-2585 effectively inhibited tumor growth when administered at 10, 20, and 40 mg/kg. At the end of treatment (day 37), ARD-2585 inhibited tumor growth by 54.9%, 74.3%, and 65.9%, respectively, compared to the blank control group. In contrast, Enzalutamide (40 mg/kg) inhibited tumor growth by 45.0%. Therefore, ARD-2585 (20 and 40 mg/kg) was more effective than Enzalutamide at 40 mg/kg in inhibiting tumor growth. Also, ARD-2585 and Enzalutamide were well tolerated and did not cause weight loss or other signs of toxicity throughout the experimental period.

Efficacy of ARD-2585 in a VCaP xenograft tumor model[3]
Pharmacodynamic (PD) data also showed that a single oral dose of ARD-2585 (20 mg/kg) was administered to mice bearing VCaP xenograft tumors. Western analysis of tumor tissue showed that ARD-2585 was effective in reducing AR protein levels at 6 and 24 hours, with a stronger effect at 24 hours.
 effects of AR degraders on AR proteins in VCaP tumors.webp")
Pharmacodynamic (PD) effects of AR degraders on AR proteins in VCaP tumors [3].
Researchers evaluated pharmacokinetic (PK) data of ARD-2585 in mice administered intravenously and orally, and ARD-2585 had an excellent volume of distribution (Vss=1.8 L/kg), long half-life (T1/2=5.5 h), and low clearance (Cl=0.3 L/h/kg) when administered intravenously at 2 mg/kg. After oral administration of ARD-2585 at 5 mg/kg, the Cmax reached 1140 ng/mL. The AUC reached 8254 h*ng/mL, and the oral bioavailability was 51%.
Finally, the researchers performed a comprehensive metabolic assay on ARD-2585 by Medicilon, including ARD-2585 metabolic stability in liver microsomes, plasma stability, and hERG assay. Stable metabolic and plasma stability of ARD-2585 in liver microsomes was demonstrated in five different species (mouse, rat, canine, monkey, and human). T1/2 >120 min. In vitro inhibition of human ERG (human ether-à-go-go-related gene) channels has been used to assess drug molecules’ potential cardiotoxicity. Researchers have conducted hERG assays with Medicilon. To test the in vitro effect of ARD-2585 on hERG potassium channel currents stably expressed in the HEK 293 cell line, using patch clamp techniques to determine the concentration-response relationship of ARD-2585 inhibition of hERG currents. ARD-2585 was found to have no inhibitory effect on hERG at concentrations up to 30 μM (the highest concentration tested).
Conclusion
The above studies can be used to develop orally active PROTAC AR reducers for the treatment of prostate cancer and provide insights and guidance for the design of orally active PROTAC reducers. Both studies utilized the Medicilon PROTAC drug discovery technology platform and preclinical DMPK evaluation system.
The Medicilon PROTAC drug discovery technology platform includes:
(1)The design of synthetic PROTAC-POI
(2)In vitro screening of PROTAC-POI
(3)The in vivo efficacy and PK/PD studies
(4)Pharmacological analysis
(5)Pharmacokinetic studies
(6)Safety evaluation of PROTAC-POI,
(7) The aggregation of experimental results and materials for IND filing to help customers accelerate the development of PROTAC-POI drugs.
References
[1]. AyeshaA Shafi, et al. Androgen receptors in hormone-dependent and castration-resistantprostate cancer. PharmacolTher. 2013 Dec;140(3):223-38.
[2]. Xin Han, et al. Strategies toward Discovery of Potent and Orally Bioavailable ProteolysisTargeting Chimera Degraders of Androgen Receptor for the Treatment of ProstateCancer. J Med Chem. 2021 Sep9;64(17):12831-12854.
[3]. Weiguo Xiang, et al. Discovery of ARD-2585 as an Exceptionally Potent and Orally Active PROTACDegrader of Androgen Receptor for the Treatment of Advanced Prostate Cancer. J Med Chem. 2021 Sep 23;64(18):13487-13509.
Medicilon PROTAC preclinical DMPK evaluation system
Through the principle of PROTAC technology, medicilon, combined with R&D cases, uses the perfect in vitro ADME and in vivo PK testing platform to establish a screening and IND evaluation system on PROTAC drugs based on small molecule compound research, focusing on drug solubility, permeability, metabolic stability, metabolite identification and in vivo PK studies, etc., to help customers quickly advance the development of PROTAC drugs.